Механизм замены лигандов. Тема работы: Элементарные стадии с участием координационных и металлоорганических соединений в растворах и на поверхности металлов и оксидов
Введение к работе
Актуальность работы . Комплексы порфиринов с металлами в высоких степенях окисления могут координировать основания гораздо более эффективно, чем комплексы М 2+ и образовывать смешанные координационные соединения, у которых в первой координационной сфере центрального атома металла наряду с макроциклическим лигандом находятся нециклические ацидолиганды, а иногда и координированные молекулы. Вопросы совместимости лигандов в таких комплексах чрезвычайно важны, так как именно в виде смешанных комплексов порфирины осуществляют свои биологические функции. Кроме того, реакции обратимого присоединения (переноса) молекул оснований, характеризующиеся умеренно высокими константами равновесия, могут успешно применяться для разделения смесей органических изомеров, для количественного анализа, для целей экологии и медицины. Поэтому исследования количественных характеристик и стехиометрии равновесий дополнительной координации на металлопорфиринах (МР) и замещения простых лигандов в них полезно не только с точки зрения теоретических познаний свойств металлопорфиринов как соединений комплексных, но и для решения практической задачи поиска рецепторов и переносчиков малых молекул или ионов. До настоящего времени систематические исследования для комплексов высокозарядных ионов металлов практически отсутствуют.
Цель работы . Настоящая работа посвящена изучению реакций смешанных порфирин-содержащих комплексов высокозарядных катионов металлов Zr IV , Hf IV , Mo V и W V с биоактивными N-основаниями: имидазолом (Im), пиридином (Py), пиразином (Pyz), бензимидазолом (BzIm), характеристике устойчивости и оптических свойств молекулярных комплексов, обоснованию ступенчатых механизмов реакций.
Научная новизна . Методами модифицированного спектрофотометрического титрования, химической кинетики, электронной и колебательной абсорбционной и 1 Н ЯМР спектроскопии впервые получены термодинамические характеристики и обоснованы стехиометрические механизмы реакций N-оснований с металлопорфиринами со смешанной координационной сферой (Х) n-2 МТРР (Х – ацидолиганд Cl - , OH - , O 2- , ТРР - дианион тетрафенилпорфирина). Установлено, что в подавляющем большинстве случаев процессы образования супрамолекул металлопорфирин – основание протекают ступенчато и включает несколько обратимых и медленных необратимых элементарных реакций координации молекул оснований и замещения ацидолигандов. Для каждой из стадий ступенчатых реакций определены стехиометрия, константы равновесий или скорости, порядки медленных реакций по основанию, спектрально охарактеризованы продукты (УФ, видимые спектры для промежуточных продуктов и УФ, видимых и ИК – для конечных). Впервые получены корреляционные уравнения, позволяющие прогнозировать устойчивость супрамолекулярных комплексов с другими основаниями. Уравнения использованы в работе для обсуждения детального механизма замещения ОН - в комплексах Mo и W на молекулу основания. Описаны свойства МР, обуславливающие перспективу использования для детектирования, разделения и количественного анализа биологически активных оснований, такие как умеренно высокая устойчивость супрамолекулярных комплексов, четкий и быстрый оптический отклик, низкий порог чувствительности, секундное время обращения.
Практическая значимость работы . Количественные результаты и обоснование стехиометрических механизмов реакций молекулярного комплексообразования имеют существенное значение для координационной химии макрогетероциклических лигандов. В диссертационной работе показано, что смешанные порфирин-содержащие комплексы проявляют высокую чувствительность и селективность в отношении биоактивных органических оснований, в течение нескольких секунд или минут дают оптический отклик, пригодный для практического детектирования реакций с основаниями - VOCs, компонентами лекарств и пищевых продуктов, благодаря чему рекомендованы для использования в качестве компонентов сенсоров оснований в экологии, пищевой промышленности, медицине и сельском хозяйстве.
Апробация работы . Результаты работы докладывались и обсуждались на:
IX Международной конференции по проблемам сольватации и комплексообразования в растворах, Плес, 2004; XII Симпозиуме по межмолекулярному взаимодействию и конформациям молекул, Пущино, 2004; XXV, XXVI и XXIX научных сессиях Российского семинара по химии порфиринов и их аналогов, Иваново, 2004 и 2006; VI Школе-конференции молодых ученых стран СНГ по химии порфиринов и родственных соединений, Санкт-Петербург, 2005; VIII научной школе - конференции по органической химии, Казань, 2005; Всероссийской научной конференции «Природные макроциклические соединения и их синтетические аналоги», Сыктывкар, 2007; XVI Международной конференции по химической термодинамике в России, Суздаль, 2007; XXIII Международной Чугаевской конференции по координационной химии, Одесса, 2007; International Conference on Porphyrins and Phtalocyanines ISPP-5, 2008; 38th International Conference on Coordination Chemistry, Israel, 2008.
Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Yс комплексом – происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев – замещение лиганда LрастворителемS, который далее легко замещается молекулой субстратаY
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
где k S иk Y – константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандомY. Например,


Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий – отщепления Cl – , присоединенияY и отщепления молекулыH 2 O.
В плоских квадратных комплексах переходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым – влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
H 2 O~NH 3 Наличие кинетического транс-эффекта и
термодинамического транс-влияния
объясняет возможность синтеза инертных
изомерных комплексов Pt(NH 3) 2 Cl 2: Реакции электрофильного замещения
(S E)
водорода металлом в координационной
сфере металла и обратные им процессы SH – H 2 O,
ROH, RNH 2 ,
RSH, ArH, RCCН. Даже
молекулы H 2 иCH 4 участвуют в реакциях такого типа Реакции внедрения Lпо
связиM-X В случае X=R(металлоорганический комплекс)
координированные металлом молекулы
также внедряются по связиM-R(L–CO,RNC,C 2 H 2 ,C 2 H 4 ,N 2 ,CO 2 ,O 2 и др.). Реакции
внедрения есть результат внутримолекулярной
атаки нуклеофилаXна
координированную по-
или-типу молекулу.
Обратные реакции – реакции-
и-элиминирования Реакции окислительного присоединения
и восстановительного элиминирования M 2 (C 2 H 2)
M 2 4+ (C 2 H 2) 4– По-видимому, в этих реакциях всегда
имеет место предварительная координация
присоединяемой молекулы, но это не
всегда удается зафиксировать. Поэтому
наличие свободного места в координационной
сфере или места, связанного с растворителем,
который легко замещается субстратом,
является важным фактором, влияющим на
реакционную способность металлокомплексов.
Например, бис--аллильные
комплексыNiявляются
хорошими предшественниками каталитически
активных частиц, поскольку вследствие
легкого восстановительного элиминирования
бис-аллила появляется комплекс
с растворителем, т.н. “голый” никель.
Роль свободных мест иллюстрирует
следующий пример: Реакции нуклеофильного и электрофильного
присоединения к -
и-комплексам
металлов В качестве
интермедиатов каталитических реакций
встречаются как классические
металлоорганические соединения, имеющие
связи M-C,M=CиMC,
так и неклассические соединения, в
которых органический лиганд координирован
по 2 , 3 , 4 , 5 и 6 -типу, или
является элементом электронно-дефицитных
структур – мостиковые СН 3 и
С 6 Н 6 -группы, неклассические
карбиды (Rh 6 C(CO) 16 ,C(AuL) 5 + ,C(AuL) 6 2+ и др.). Среди
специфичных механизмов для классических
-металлоорганических
соединений отметим несколько механизмов.
Так, установлено 5 механизмов электрофильного
замещения атома металла по связиM-C. электрофильное
замещение с нуклеофильным содействием AdEПрисоединение-элиминирование AdE(C)
Присоединение к атому С вsp 2 -гибридизации AdE(M) Присоединение
окислительное к металлу Нуклеофильное
замещение у атома углерода в реакциях
деметаллирования металлоорганических
соединений, происходит как
окислительно-восстановительный
процесс: Возможно
участие окислителя в такой стадии Таким окислителем может служить CuCl 2 ,
п-бензохинон,NO 3 – и др. соединения. Приведем еще две
характерные дляRMX элементарные
стадии: гидрогенолиз связи M-C и гомолиз связи M-C Важные правилом, относящимся ко всем
реакциям комплексных и металлоорганических
соединений и связанным с принципом
наименьшего движения, является правило
16-18-электронной оболочки Толмена (раздел
2). Реакции координационных соединений всегда происходят в координационной сфере металла со связанными в ней лигандами. Поэтому очевидно, что для того, чтобы вообще что-то происходило, лиганды должны уметь в эту сферу попадать. Это может происходить двумя способами: С первым способом мы уже ознакомились, когда обсуждали координационную ненасыщенность и 18-электронное правило. Вторым займемся здесь. Но обычно действует негласное правило – количество занятых координационных мест не изменяется. Иными словами, при замещении не меняется счет электронов. Замещение лиганда одного типа на другой вполне возможно и часто происходит в реальности. Обратим только внимание на корректное обращение с зарядами, когда меняется L-лиганд на X-лиганд и наоборот. Если мы про это забудем, то изменится степень окисления металла, а замещение лигандов не является окислительно-восстановительным процессом (если найдете или придумаете противный пример, дайте знать – зачет автоматом сразу, если я не смогу доказать, что вы ошиблись, впочем даже и в этом случае гарантирую положительный вклад в карму) . С более сложными лигандами сложностей не больше – нужно просто помнить довольно очевидное правило: количество лигандных мест (то есть общее количество лигандов или лигандных центров X- или L-типов) сохраняется. Это непосредственно следует из сохранения счета электронов. Вот самоочевидные примеры. Обратим внимание на последний пример. Исходный реагент для этой реакции дихлорид железа FeCl 2 . Еще недавно мы бы сказали: “Это просто соль, причем тут координационная химия?”. Но больше мы не будем себе позволять такое невежество. В химии переходных металлов не бывает “просто солей”, любые производные суть координационные соединения, к которым применимы все рассуждения про счет электронов, d-конфигурацию, координационную насыщенность и т.п. Дихлорид железа, так как мы его привыкли писать, оказался бы комплексом Fe(2+) типа MX 2 с конфигурацией d 6 и числом электронов 10. Маловато что-то! Нормально? Ведь мы уже разобрались, что лиганды бывают неявные. Чтобы сделать реакцию, нам нужен растворитель, и для таких реакций это, скорее всего, ТГФ. Растворение кристаллической соли железа в ТГФ и происходит именно потому, что донорный растворитель занимает свободные места, и энергия этого процесса компенсирует разрушение кристаллической решетки. Мы не смогли бы растворить эту “соль” в растворителе, не предоставляющем услуг сольватации металла за счет основности Льюиса. В данном случае, и в миллионе подобных, сольватация это просто координационное взаимодействие. Напишем, просто для определенности результат сольватации в виде комплекса FeX 2 L 4 , у которого два иона хлора остаются в координационной сфере в виде двух X-лигандов, хотя скорее всего они тоже вытеснены молекулами донорного растворителя с образованием заряженного комплекса FeL 6 2+
. В данном случае это не так важно. И так, и эдак мы можем спокойно считать, что у нас 18-электронный комплекс и слева, и справа. Если мы помним органическую химию, то там было два механизма замещения при насыщенном атоме углерода – SN1 и SN2. В первом замещение происходило двухстадийно: старый заместитель сначала уходил, оставляя вакантную орбиталь на атоме углерода, на которую следом заходил новый заместитель с парой электронов. Второй механизм предполагал, что уход и приход осуществляются одновременно, согласованно, а процесс был одностадийным. В химии координационных соединений вполне можно представить нечто похожее. Но появляется и третья возможность, которой не было у насыщенного атома углерода – сначала присоединяем новый лиганд, потом отцепляем старый. Сразу становится понятно, что этот третий вариант вряд ли возможен, если комплекс уже имеет 18 электронов и является координационно насыщенным. Но вполне возможен, если число элетронов 16 или меньше, то есть комплекс ненасыщен. Тут же вспомним и очевидную аналогию из органической химии – нуклеофильное замещение у ненасыщенного атома углерода (в ароматическом кольце или у карбонильного углерода) тоже идут сначала как присоединение нового нуклеофила, и потом отщепление старого. Итак, если у нас 18 электронов, то замещение идет как отщепление-присоединение (любители “умных” словечек используют термин диссоциативно-ассоциативный или просто диссоциативный механизм). Другой путь потребовал бы расширения координационной сферы до счета в 20 электронов. Это не является абсолютно невозможным, и такие варианты иногда даже рассматриваются, но это точно очень невыгодно и каждый раз в случае подозрения на такой путь требуются очень весомые доказательства. В большинстве таких историй исследователи в конце концов приходили к выводу, что они что-то просмотрели или не учли, и ассоциативный механизм отвергался. Итак, если исходный комплекс с 18 электронами, то сначала один лиганд должен уйти, затем на его место прийти новый, например: Если мы хотим ввести в координационную сферу гапто-лиганд, занимающий несколько мест, то сначала мы должны их все освободить. Как правило, это происходит только в достаточно жестких условиях, например, чтобы в карбониле хрома заменить три карбонила на η 6 -бензол, смесь много часов нагревают под давлением, время от времени стравливая высвободившийся оксид углерода. Хотя в схеме нарисована диссоциация трех лигандов с образованием очень ненасыщенного комплекса с 12 электронами, в реальности реакция, скорее всего происходит стадийно, уходит по одному карбонилу, а бензол заходит в сферу, постепенно увеличивая гаптность, через стадии минус CO – дигапто – минус еще одна CO – тетрагапто – минус еще одна CO – гексагапто, так чтобы меньше 16 электронов не получалось. Итак, если у нас комплекс с 16 электронами или меньше, то замещение лиганда, скорее всего, идет как присоединение-отщепление (для любителей глубокомысленных слов: ассоциативно-диссоциативный или просто ассоциативный): новый лиганд сначала приходит, затем старый уходит. Напрашиваются два очевидных вопроса: почему уходит старый лиганд, ведь 18 электронов это очень хорошо, и почему бы и в этом случает не сделать наоборот, как в 18-электронных комплексах. На первый вопрос ответить легко: у каждого металла свои привычки, и некоторые металлы, особенно из поздних, с почти полностью заполненными d-оболочками, предпочитают 16-электронный счет и соответствующие структурные типы, поэтому и выбрасывают лишний лиганд, возвращаясь к любимой конфигурации. Иногда в дело еще вмешивается пространтсвенный фактор, уже имеющиеся лиганды большие и дополнительный чувствует себя, как пассажир автобуса в час пик. Проще сойти и прогуляться пешком, чем так мучиться. Впрочем, можно выпихнуть другого пассажира, пусть погуляет, а мы поедем. Второй вопрос тоже прост – в этом случае диссоциативный механизм должен был бы сначала дать 14-электронный комплекс, а это редко бывает выгодно. Вот пример. Для разнообразия заменим X-лиганд на L-лиганд, и не запутаемся в степенях окисления и зарядах. Еще раз: при замещении степень окисления не меняется, и если ушел X-лиганд, то потерю нужно скомпенсировать зарядом на металле. Если про это забыть, то степень окисления уменьшилась бы на 1, а это неверно. И еще одна странность. Образовалась связь металл-пиридин за счет неподеленной пары на азоте. В органической химии в этом случае мы обязательно показали бы плюс на азоте пиридина (например, при протонировании или образовании четвертичной соли), но мы никогда не делаем это в координационной химии ни с пиридином, ни с любыми другими L-лигандами. Это страшно раздражает всех, кто привык к строгой и недвусмысленной системе рисования структур в органической химии, но придется привыкать, это не так сложно. А точного аналога SN2 в химии координационных соединений нет, есть далекий, но он относительно редок и нам не очень нужен.
Про механизмы замещения лигандов можно было бы вообще не говорить, если бы не одно чрезвычайно важное обстоятельство, которым мы будем очень много пользоваться: замещение лигандов, хоть ассоциативное, хоть диссоциативное обязательно предполагает диссоциацию старого лиганда. И нам очень важно знать, какие лиганды легко уходят, а какие уходят плохо, предпочитая оставаться в координационной сфере металла. Как мы скоро увидим, в любой реакции часть лигандов остается в координационной сфере и не изменяется. Такие лиганды принято называть лигандами-зрителями (если не хотите таких простых, “ненаучных” слов, используйте английское слово spectator в местной транскрипции спектатор, лиганд-спектатор, только, умоляю, не спектейтор – это невыносимо!). А часть непосредственно участвует в реакции, превращаясь в продукты реакции. Такие лиганды называют акторами (не актерами!), то есть действующими. Вполне понятно, что лиганды-акторы нужно в координационную сферу металла легко вводить и выводить, иначе реакция просто застрянет. А вот лиганды-спектаторы лучше в координационной сфере оставлять по многим причинам, но хотя бы и по такой банальной как необходимость избежать излишней суеты вокруг металла. Лучше, чтобы только лиганды акторы и в необходимых количествах могли участвовать в нужном процессе. Если доступных координационных мест будет больше, чем необходимо, на них могут усесться лишние лиганды-акторы, и даже такие, которые будут участвовать в побочных реакциях, снижая выход целевого продукта и селективность. Лиганды-спектаторы кроме того почти всегда осуществляют множество важных функций, например, обеспечивают растворимость комплексов, стабилизируют правильное валентное состояние металла, особенно если оно не совсем обычное, помогают отдельным стадиям, обеспечивают стереоселективность, и т.п. Пока не расшифровываем, потому что все это мы будем обсуждать подробно, когда доберемся до конкретных реакций. Получается, что часть лигандов в координационной сфере должна быть прочно связанной и не склонной к диссоциации и замещению другими лигандами. Такие лиганды принято называть координационно стабильными
. Или просто стабильными, если из контекста ясно, что речь идет о прочности связи лигандов, а не об их собственной термодинамической стабильности, которая как раз нас совершенно не волнует. А лиганды, которые легко и охотно входят и выходят, и всегда готовы уступить место другим, называют координационно лабильными
, или просто лабильными, и здесь, к счастью, никаких двусмысленностей нет. Вот, наверное, самый яркий пример того, что в координационной сфере очень нестабильная молекула может стать отличным лигандом, причем по определению координационно стабильным, хотя бы потому, что если она рискнет выйти из теплой и уютной сферы наружу, ничего хорошего ее не ждет (ценой выхода будет как раз энергия антиароматической дестабилизации). Циклобутадиен и его производные – самые известные примеры антиароматичности. Эти молекулы существуют только при низких температурах, и в сильно искаженном виде, – чтобы уйти как можно дальше от антиароматичности, цикл искажается в вытянутый прямоугольник, снимая делокализацию и максимально ослабляя сопряжение двойных связей (по другому это называется эффектом Яна-Теллера 2 рода: вырожденная система, а циклобутадиен-квадрат представляет собой вырожденный бирадикал, вспомните круг Фроста, – искажается и снижает симметрию, чтобы снять вырождение). Но в комплексах циклобутадиен и замещенные циклобутадиены – отличные тетрагапто-лиганды, и геометрия таких лигандов – именно квадрат, с одинаковыми длинами связей. Как и почему это происходит – отдельная история, и далеко не такая очевидная, какой ее часто подают. Нужно понимать, что железобетонного забора с колючей проволокой и вышками охраны между областями лабильных и стабильных лигандов нет. Во-первых, это зависит от металла, и в этом контексте неплохо работает ЖМКО. Например, поздние переходные металлы предпочитают мягкие лиганды, а ранние – жесткие. Скажем, иодид очень крепко держится за d 8 атомы палладия или платины, но редко вообще входят в координационную сферу титана или циркония в конфигурации d 0 . Но во многих комплексах металлов с не столь ярко выраженными признаками, иодид проявляет себя как вполне лабильный лиганд, легко уступающий место другим. При прочих равных условиях: Повторим все еще раз, только с другой стороны В координационной сфере металлов сохраняются (являются координационно стабильными) как правило: Последнее условие выглядит странно, но представьте себе комплекс, у которого много разных лигандов, среди которых нет безусловно стабильных (нет хелаторов и полигапто-лигандов). Тогда в реакциях лиганды будут меняться, условно говоря, в порядке относительной лабильности. Наименее лабильный и останется последним. Такой фокус имеет место, например, когда мы используем фосфиновые комплексы палладия. Фосфины – относительно стабильные лиганды, но, когда их много, а металл богат электронами (d 8 , d 10), они уступают, один за другим, место лигандам-акторам. Но последний фосфиновый лиганд обычно остается в координационной сфере, и это очень хорошо с точки зрения тех реакций, в которых эти комплексы участвуют. Мы еще вернемся к этой важной проблеме. Вот довольно типичный пример, когда от исходной координационной сферы фосфинового комплекса палладия в реакции Хека остается только один, “последний” фосфин. Этот пример очень близко подводит нас к важнейшей концепции в реакциях комплексов переходных металлов – концепции контролирующего лиганда (ligand control). Обсудим позднее. При замещении одних лигандов на другие важно не переборщить с реакционной способностью входящего лиганда. Когда мы имеем дело с реакциями органических молекул, нам важно доставить в координационную сферу ровно по одной молекуле каждого из реагентов. Если вместо одной войдет две молекулы, высока вероятность побочных реакций с участием двух одинаковых лигандов. Возможна также и потеря реакционной способности из-за насыщения координационной сферы и невозможности введения в нее других необходимых для ожидавшегося процесса лигандов. особенно часто эта проблема возникает при введении в координационную сферу сильных анионных нуклеофилов, например, карбанионов. Чтобы избежать этого, используют менее реакционноспособные производные, в которых, вместо катиона щелочного металла, обусловливающего высокую ионность связи, используют менее электроположительные металлы и металлоиды (цинк, олово, бор, кремний, и т.п.), образующие ковалентные связи с нуклеофильной частью. Реакции таких производных с производными переходных металлов дают продукты замещения лигандов, в принципе, точно так же как если бы нуклеофил был в анионной форме, но из-за сниженной нуклеофильности с меньшими осложнениями и без побочных реакций. Такие реакции замещения лигандов принято называть переметаллированием (transmetallation), чтобы подчеркнуть то очевидное обсоятельство, что нуклеофил как будто бы меняет металлы – более электроположительный на менее электроположительный. В этом названии, таким образом, заложен элемент малоприятной шизофрении – мы вроде бы уже договорились, что будем на все реакции смотреть с точки зрения переходного металла, но вдруг опять сорвались и смотрим на эту реакцию и только на эту реакцию с точки зрения нуклеофила. Придется потерпеть, так сложилась терминология и так принято. На самом деле, это слово восходит к ранней химии металлоорганических соединений и к тому, что действие литий или магнийорганических соединений на галогениды разных металлов и металлоидов – один из основных методов синтеза всякой металлоорганики, в первую очередь непереходной, и реакция, которую мы сейчас рассматриваем в химии координационных соединений переходных металлов – просто обобщение старинного метода металлоорганической химии, из которого она вся и выросла. Переметаллирование и похоже на обычное замещение, и не похоже. Похоже – если мы считаем непереходный металлоорганический реагент просто карбанионом с противоионом, то есть связь углерод-непереходный металл ионной. Но это представление похоже на правду только для самых электроположительных металлов – для магния. Но уже для цинка и олова это представление очень далеко от истины. Поэтому в реакцию вступают две σ-связи и четыре атома на их концах. В результате образуются две новые σ-связи и четыре атома связываются друг с другом в другом порядке. Скорее всего, все это происходит одновременно в четырехчленном переходном состоянии, и сама реакция имеет согласованный характер, как и очень многие другие реакции переходных металлов. Обилие электронов и орбиталей буквально на все вкусы и все виды симметрий делает переходные металлы способными одновременно поддерживать связи в переходных состояниях с несколькими атомами. В случае переметаллирвоания получаем частный случай очень общего процесса, который называется просто метатезисом σ-связей (σ-bond metathesis). Не путайте только с настоящими метатезисами олефинов и ацетиленов, которые являются полноценными каталитическими реакциями со своими механизмами. В данном случае речь идет о механизме переметаллирования или другого процесса, в котором происходит нечто подобное. Комплексные соединения. Их строение на основе координационной теории А. Вернера. Комплексный ион, его заряд. Катионные, анионные, нейтральные комплексы. Номенклатура, примеры. Реакции замещения лигандов. Константа нестойкости комплексного иона, константа устойчивости.
К уст= 1/ К нест (обратная величина) 43.Конкуренция за лиганд или за комплексообразователь: изолированное и совмещенное равновесия замещения лигандов. Общая константа совмещенного равновесия замещения лигандов.
В результате конкуренции протон разрушает достаточно прочный комплекс, образуя слабо диссоциирующее вещество - воду. Cl + NiS0 4 +4NH 3 ^ S0 4 +AgCl I Это уже пример конкуренции лиганда за комплексообразователь, с образованием более прочного комплекса (K H + =9,3-1(Г 8 ; К Н [М(Ш 3) 6 ] 2+ = 1,9-Ю -9) и труднорастворимого соединения AgCl - K s = 1,8 10" 10 Представления о строении металлоферментов и других биокомплексных соединений (гемоглобин, цитохромы, кобаламины). Физико-химические принципы транспорта кислорода гемоглобином Кобаламины. Витами́нами B 12
называют группу кобальтсодержащих биологически активных веществ, называемых кобаламинами. К ним относят собственно цианокобаламин
, гидроксикобаламин и две коферментные формы витамина B 12: метилкобаламин и 5-дезоксиаденозилкобаламин. Иногда в более узком смысле витамином B 12 называют цианокобаламин, так как именно в этой форме в организм человека поступает основное количество витамина B 12 , не упуская из вида то, что он не синоним с B 12 , и несколько других соединений также обладают B 12 -витаминной активностью . Витамин B 12 также называется внешним фактором Касла. B 12 имеет самую сложную по сравнению с другими витаминами химическую структуру, основой которой является корриновоекольцо. Коррин во многом похож на порфирин (сложная химическая структура, входящая в состав гема, хлорофилла ицитохромов), но отличается от порфирина тем, что два пиррольных цикла в составе коррина соединены между собой непосредственно, а не метиленовым мостиком. В центре корриновой структуры располагается ион кобальта. Четыре координационных связи кобальт образует с атомами азота. Ещё одна координационная связь соединяет кобальт сдиметилбензимидазольным нуклеотидом. Последняя, шестая координационная связь кобальта остаётся свободной: именно по этой связи и присоединяется цианогруппа, гидроксильная группа, метильный или 5"-дезоксиаденозильный остаток с образованием четырёх вариантов витамина B 12 , соответственно. Ковалентная связь углерод-кобальт в структуре цианокобаламина - единственный известный в живой природе пример ковалентной связи переходный металл-углерод. Лиганды - ионы или молекулы, которые непосредственно связаны с комплексообразователем и являются донорами электронных пар. Эти электроноизбыточные системы, имеющие свободные и подвижные электронные пары, могут быть донорами электронов, например: Соединения р-элементов проявляют комплексообразующие свойства и выступают в комплексном соединении в качестве лигандов. Лигандами могут быть атомы и молекулы (белка, аминокислот, нуклеиновых кислот, углеводов). Эффективность и прочность донорно-акцкпторного взаимодействия лиганда и комплексообразователя определяется их поляризуемостью-способностью частицы трансформировать свои электронные оболочки под внешним воздействием. Кнест= 2 / К уст=1/Кнест Реакции замещения лигандов Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Yс комплексом – происходит по трем механизмам: а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса Суть процесса в большинстве случаев – замещение лиганда LрастворителемS, который далее легко замещается молекулой субстратаY б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда в) Синхронное замещение (типа S N 2) без образования интермедиата Представления о строении металлоферментов и других биокомплексных соединений (гемоглобин, цитохромы, кобаламины). Физико-химические принципы транспорта кислорода гемоглобином. Особенности строения металлоферментов.
Биокомплексные соединения значительно различаются по устойчивости. Роль металла в таких комплексах высокоспецифична: замена его даже на близкий по свойствам элемент приводит к значительной или полной утрате физиологической активности. 1.
В12: содержит 4 пиррольных кольца,ион кобальта и группы CN-. Способствует переносу атома H на атом С в обмен на какую либо группу, участвует в процессе образования дезоксирибозы из рибозы. 2.
гемоглобин:имеет четвертичную структуру. Четыре полипептидные цепи, соединённые вместе, образуют почти правильную форму шара, где каждая цепь контактирует с двумя цепями. Гемоглобин
- дыхательный пигмент, придающий крови красный цвет. Гемоглобин состоит из белка и железопорфирина и переносит кислород от органов дыхания к тканям тела и углекислый газ от них к дыхательным органам. Физико-химические принципы транспорта кислорода гемоглобином
- Атом (Fe (II)) (один из компонентов гемоглобина) способен образовывать 6 координационных связей. Из них четыре используются для закрепления самого атома Fe(II) в геме, пятая связь - для связывания гема с белковой субъединицей, а с помощью шестой связи происходит связывание молекулы О 2 или СО 2. Металло-лигандный гомеостаз и причины его нарушения. Механизм токсического действия тяжелых металлов и мышьяка на основе теории жестких и мягких кислот и оснований (ЖМКО). Термодинамические принципы хелатотерапии. Механизм цитотоксического действия соединений платины. В организме непрерывно происходит образование и разрушение биокомплексов из катионов металлов и биолигандов (порфинов, аминокислот, белков, полинуклеотидов), в состав которых входят донорные атомы кислорода, азота, серы. Обмен с окружающей средой поддерживает концентрации этих веществ на постоянном уровне, обеспечивая металло-лигандный гомеостаз
.
Нарушение сложившегося равновесия ведет к ряду патологических явлений – металлоизбыточным и металлодефицитным состояниям. В качестве примера можно привести неполный перечень заболеваний, связанных с изменением металло-лигандного баланса только для одного иона – катиона меди. Дефицит этого элемента в организме вызывает синдром Менкеса, синдром Морфана, болезнь Вильсона-Коновалова, цирроз печени, эмфизему лёгких, аорто- и артериопатии, анемии. Избыточное поступление катиона может вести к серии заболеваний самых разных органов: ревматизму, бронхиальной астме, воспалению почек и печени, инфаркту миокарда и т.д., называемых гиперкупремиями. Известен и профессиональный гиперкупреоз – медная лихорадка. Циркуляция тяжелых металлов происходит частично в виде ионов или комплексов с аминокислотами, жирными кислотами. Однако ведущая роль в транспорте тяжелых металлов принадлежит белкам, образующим с ними прочную связь. Они фиксируются на клеточных оболочках, блокируют тиоловые группы мембранных протеинов
– 50% из них белки-ферменты, нарушают стабильность белково-липидных комплексов клеточной оболочки и ее проницаемость, вызывая выход из клетки калия и проникновение в нее натрия и воды. Подобное действие этих ядов, активно фиксирующихся на красных кровяных клетках, приводит к нарушению целостности мембран эритроцитов, торможению в них процессов аэробного гликолиза и метаболизма вообще и накоплению гемолитически активной перекиси водорода вследствие торможения пероксидазы в частности, что приводит к развитию одного из характерных симптомов отравления соединениями этой группы – к гемолизу. Распределение и депонирование тяжелых металлов и мышьяка происходят практически во всех органах. Особый интерес представляет способность этих веществ накапливаться в почках, что объясняется богатым содержанием в почечной ткани тиоловых групп, наличием в ней белка – металлобионина, содержащего большое количество тиоловых групп, что способствует длительному депонированию ядов. Высокой степенью накопления токсических соединений этой группы отличается и ткань печени, также богатая тиоловыми группами и содержащая металлобионин. Срок депонирования, например, ртути может достигать 2 мес и более. Выделение тяжелых металлов и мышьяка происходит в разных пропорциях через почки, печень (с желчью), слизистую оболочку желудка и кишечника (с калом), потовые и слюнные железы, легкие, что сопровождается, как правило, поражением выделительных аппаратов этих органов и проявляется соответствующей клинической симптоматикой. Смертельная доза для растворимых соединений ртути 0,5 г, для каломели 1–2 г, для медного купороса 10 г, для ацетата свинца 50 г, для свинцовых белил 20 г, для мышьяка 0,1–0,2 г. Токсической считается концентрация ртути в крови более 10 мкг/л (1γ%), в моче более 100 мкг/л (10γ%), концентрация меди в крови более 1600 мкг/л (160γ%), мышьяка более 250 мкг/л (25γ%) в моче. Хелатотерапия – это выведение токсичных частиц из организма, основанное на хелатировании их комплексонатами s–элементов. Препараты, применяемые для выведения инкорпорированных в организме токсичных частиц, называют детоксикантами.

Реакции координированных лигандов
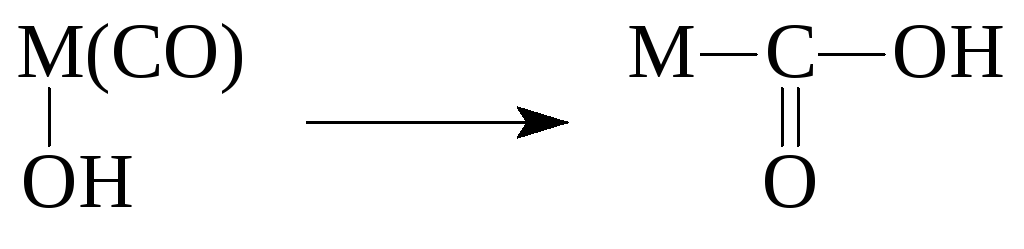









Реакции металлоорганических соединений



Замещаться могут в любых комбинациях лиганды любых типов
Замещение с участием гапто-лигандов

Замещение, присоединение и диссоциация лигандов тесно и неразрывно связаны



Стабильные и лабильные лиганды
Циклобутадиен как лиганд
Координационно лабильные лиганды
Координационно стабильные лиганды

Переметаллирование
![]()
Как происходит переметаллирование?





 К нестойкости- это отношение произведений концентрации распавшихся ионов на нераспавшееся количество.
К нестойкости- это отношение произведений концентрации распавшихся ионов на нераспавшееся количество. Вторичная диссоциация -
распад внутренней сферы комплекса на составляющие ее компоненты.
Вторичная диссоциация -
распад внутренней сферы комплекса на составляющие ее компоненты.


Константа нестойкости:
Цитохромы
- сложные белки (гемопротеиды), осуществляющие в живых клетках ступенчатый перенос электронов и/или водорода от окисляемых органических веществ к молекулярному кислороду. При этом образуется богатое энергией соединение АТФ.
Кобаламины
- природные биологически активные кобальторганические соединения. Структурной основой К. является корриновое кольцо, состоящее из 4 пиррольных ядер, у которых атомы азота связаны с центральным атомом кобальта.





